Scientific methods
Here we describe some of the techniques we use in our scientific work to help us better understand the foods we make in our culinary R&D—especially their microbial ecologies (if fermented), their chemistry, and their flavours. Many of these techniques have wider research applications, not just for microbes or foods; here we focus on how they relate to and enable our own work. The examples we include are just how we have used these methods so far. We will add even more as our research develops.
Need an introduction to or a quick refresher on transcription and translation, DNA and RNA? We’d recommend these resources for some of the science that underpins these techniques.
Table of Contents
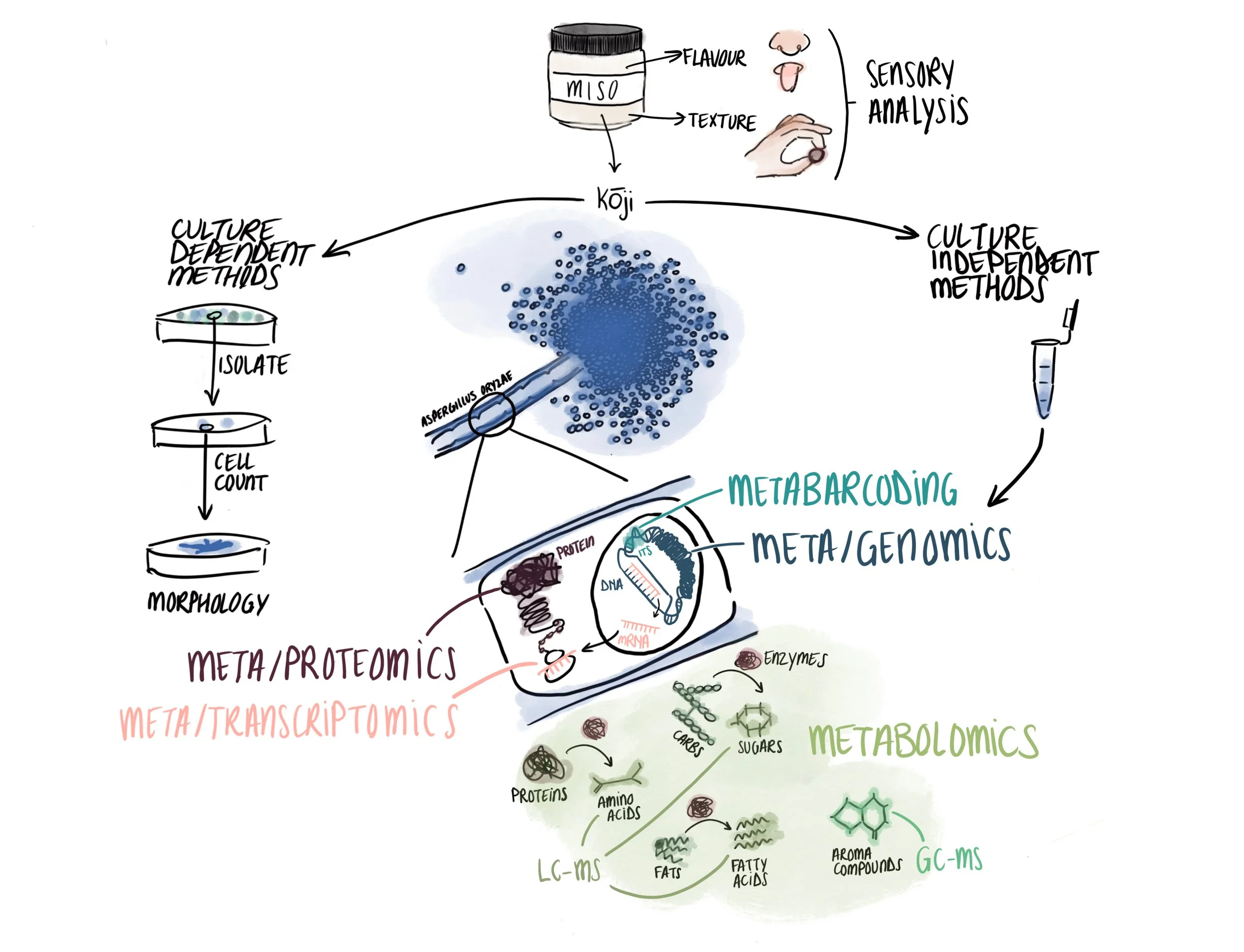
i. What scientific methods are available to us to better understand a sample of food?
Before analysing a sample of food, it is essential to first consider what exactly we’d like to understand about it. Different questions require different methods, each of which has its own merits and drawbacks.
Much of our scientific work focuses on the microbiology of novel fermentations, so that is what many of the methods we share here are for. In microbiology, there are two main groups of methods. The first comprises culture-dependent methods, which involve cultivating samples in Petri dishes. These classical methods were the backbone of microbiology for much of its history, leading to many important discoveries. They have their limits, however; since <1% of known microorganism species are cultivable in this way, these methods are only useful for a handful of well-known species, don’t allow us to study microbial communities in situ (where they actually live), and are time- and space-consuming to perform.¹ Even though other methods have risen to prominence, culture-dependent methods still have several important uses, which we discuss below.
The second group comprises culture-independent methods (aka molecular biology) like metagenomics, metabolomics, metatranscriptomics, and other ‘omics’ analyses. These methods do not require culturing microorganisms first, which can (sometimes) make analysis relatively quicker and easier.² This culture-independence opens up many possibilities for studying microbial communities in situ, including classifying microbial species within a sample and describing the potential or actual functions of the microbial ecosystem. Even so, culture-independent methods can’t entirely replace culture-dependent methods, and combining the two can often be ideal. In our work, we often use a combination of both, depending on the question we want to answer.
Beyond microbiology, other techniques, like sensory analysis, texture analysis and nutritional analysis, can give an even richer understanding of the physical, chemical, and sensory characteristics of foods.
All of these methods can be combined for especially illuminating results.
ii. What do we want to find out about our sample?
There are four main questions we can ask here: 1) Which microbes are there?; 2) What are the microbes capable of doing?; 3) What are the microbes actually doing? and 4) How do the microbes impact the food’s taste, smell, texture, and nutritional profile?

Which microbes are there?
Our story starts with DNA. Metabarcoding (aka metagenetics or amplicon sequencing) uses the polymerase chain reaction (PCR) to amplify specific target DNA regions within a sample , which can be used to identify and classify the taxa present.³ For our microbiological research, we typically amplify either the 16S rRNA gene, which is unique to bacterial species, or the ITS gene, which is unique to fungal species. The reason these genes are used is because they are particularly well-conserved within kingdoms, while also having slight variations between taxa. So all bacteria have a 16S rRNA gene, but this gene varies slightly between species, so it is a good indicator of taxonomy. Sometimes this gene is referred to as a microbial ‘fingerprint’. Taxonomic classification using metabarcoding can often only be done to the genus level, though sometimes also to the species level, depending on how much variation there is in the gene between species within the same genus. Metabarcoding is cheaper than metagenomics (see below) because it amplifies and sequences only one gene, rather than entire genomes, but for the same reason is less precise. Since 16S and ITS have to be amplified separately, it also only shows the relative abundance of fungi and of bacteria separately, rather than the combined relative abundance of all microorganisms together.
Shotgun metagenomics is a more precise way of studying the taxonomic diversity in a sample to the species level, or even lower, such as to the strain level for microbes. It uses fragments of all DNA in a sample, which, when sequenced and assigned to taxa in databases gives the combined relative proportion of species within the whole sample as well as their associated genes and potential functions. We use this technique in much of our research to precisely determine the taxonomic composition of microbes in our samples. While metagenomics is generally more powerful than metabarcoding, it is not always the right option. Sometimes we want to sequence only the bacterial and/or fungal species in a sample, and not the plant or animal DNA, for example. In this case metabarcoding is useful because we can target just the kingdoms we want, and not spend sequencing power (and $$) on sequencing DNA we don’t need.
Classifying microbes and describing microbial community composition, whether through metabarcoding or metagenomics, has several uses in our research, including:
revealing how different substrates shape the composition of microbial communities, such as in our work with novel misos;
investigating how the microbial ecologies of novel fermentations like endive root tonic and novel misos might consist of new assemblages of microorganisms not previously found elsewhere, or might be strikingly similar to traditional versions even if made totally differently, as in our work with spontaneous kombuchas;
studying how different environments shape microbial communities and potentially lineages, as in our work with the space miso or kōji evolution;
linking the presence of specific microbes with specific flavours and other food characteristics, an ongoing interest we are pursuing in many projects; and
determining the safety of a novel food by revealing if it contains any potential pathogens and/or toxigenic genes, an approach we’ve used in our novel miso work.
After identifying species of microbes using these methods, culture-dependent methods can then be used to isolate, culture and describe species present in a sample. This can be especially helpful when trying to describe potentially new species or strains, as we did with a possible new species of Exiguobacterium in our work on novel misos.

What are the microbes capable of doing?
DNA can tell us not only who is there, but also what they are capable of. Metagenomics can also be used, in combination with bioinformatic techniques, to reconstruct entire genomes present in the sample, called Metagenome-Assembled Genomes (MAGs), to analyse the potential function of all the genes present. We used this technique in our work on novel misos, to get a better sense of what some of the species were capable of in the miso environment, and also to study the strain adaptation of Staphylococcus epidermidis, typically associated with the human skin microbiome, to some of the misos. We will also likely be using it in our ongoing analysis from our kōji evolution experiment, reconstructing Aspergillus oryzae genomes from kōji metagenomes (kōji being a community with bacteria and yeasts as well) to study how they may have changed when grown in different culinary environments.
Genomics can help us understand the genome of single species more directly, and with even greater precision, using culture-dependent methods to isolate and sequence the DNA of just this species. We used genomics (in combination with culture-dependent techniques) in our space miso project, in which we isolated A. oryzae from different samples of miso that were fermented in Denmark, the USA and on the International Space Station, so we could extract just this species’ DNA. We then studied their genomes to assess the types of mutations caused by the space environment and their impact on gene functioning. We might also use this method in our kōji evolution study, if needed, for more precise genomic information to supplement the MAGs.
Whilst metagenomics can reveal the overall genetic composition of a sample, it cannot differentiate between cells that are dead or alive, as it only analyses DNA, regardless of where it is found. Culture-dependent methods can again help here: after cultivating the microbes in a sample, plate counting can be conducted to estimate the number of viable, or living, colony-forming units (CFUs) in a sample. This is a fairly straightforward method—it involves simply counting the dots on the plate by hand. Flow cytometry is another technique that can be used to measure the proportion of active cells in a sample. The abundance of active microbes determines, for example, the concentrations of the metabolites that they produce, which in turn determine fermented food characteristics. For example, in 3-month-old miso, despite the essential role of A. oryzae (in the form of kōji) in the early stages of miso production, plate counting reveals that it doesn’t appear to be active in large quantities after 3 months, even though the relatively high abundance of its DNA in a sample might suggest otherwise. This is a good example of why it is so important to critically interpret and contextualise one’s data. In some of our novel miso data, for example, we had large amounts of A. oryzae reads, and if we didn’t know about the microbiological and culinary context of miso, we might then conclude that there was lots of A. oryzae actually living in the finished miso. Integrating different kinds of data helps tell a fuller story.

What are the microbes actually doing?
Since genes are expressed differently depending on environmental and other factors, knowing what genes are present doesn’t necessarily mean that they are active. This is where we need to study transcription—which genes are activated and create RNA. Metatranscriptomics can be used to determine all the genes that are expressed within a sample based on extracted RNA, whilst transcriptomics can do the same for a single species (comparable to the difference between metagenomics and genomics for DNA). Genes may be expressed differently depending on how an organism adapts to environmental factors like temperature, pH, humidity and nutrient availability. Microbes may adapt to their environment by generating compounds like flavour precursors and toxins to outcompete other microbes, which can impact the physical, sensory, and safety characteristics of foods.
Metatranscriptomics and transcriptomics allow us to understand the behaviour of different microorganisms in different conditions, which can then help us create the best conditions for the characteristics we want a food to have. For example, Penicillium camemberti is a fungus used to produce ripened Camembert-style cheeses with characteristic bloomy rinds, and is a good candidate for making plant cheeses in this style as it produces a strong cheesy aroma. We used transcriptomics to understand how P. camemberti adapts to different environments—like cow’s milk and different plant substrates—by revealing how genes are transcribed and expressed in each environment. We could also use this method to similarly study how microbes modulate their gene expression to adapt to different novel misos. We plan to do more of this kind of analysis in the future to study how different microbes adapt their behaviour to novel substrates.
From transcription we move to translation—how RNA codes for the synthesis of proteins. Metaproteomics can be used to detect the presence and abundance of different types of proteins under specific conditions, which can reveal important information about foods’ structure. (Proteomics does this for a single species.) For example, casein is a protein that can be coagulated with the addition of an enzyme and is responsible for many of the unique structural properties of cheeses made from animal milk. Metaproteomics could inform the design of plant cheeses by revealing all the proteins present within plant ingredients, helping us to understand their structure better and improve processes to create delicious, appropriately-textured cheeses using those ingredients. In a similar vein, it could also be used to screen a range of microbes for new enzymes that might coagulate plant milks better, creating the stretchy, melty, gooey textures so beloved in dairy cheese. Furthermore, it might also be used to understand foods’ umami potential. We haven’t used this method so much yet, but we plan to.
Proteins and enzymes (which are types of proteins) bring us to metabolism more generally, the transformation of molecules within the cell for its proper functioning. Metabolomics can be used to characterise and quantify the different molecules (or metabolites) that are present in a sample, like carbohydrates, amino acids, lipids, vitamins and minerals, which all impact food characteristics like flavour, texture and nutrition. In fermented foods, these methods can also indicate the different metabolites that microorganisms are producing and/or consuming. Liquid chromatography and mass spectrometry (LC-MS) can be used together to detect and identify small, non-volatile compounds like amino acids, which impact the growth of different microorganisms, short-chain fatty acids which impact the texture and mouthfeel of foods, and potentially dangerous mycotoxins. Gas chromatography and mass spectrometry (GC-MS) can be used together to detect and identify volatile compounds, like aroma-generating compounds. We’ve used metabolomics in multiple projects: in our space miso project to characterise the volatile molecules, amino acids, and organic acids of the misos; in our plant cheese research to reveal differences in amino acids and short-chain fatty acids between soy and dairy cheese models; in our R&D work with tempe to investigate and quantify its chickeny flavours under certain growth conditions; and in a collaboration investigating the flavour chemistry and sensory profiles of insect frass tea.
Culture-dependent methods can be used to assess the morphology of different microbes—i.e. what they look like when cultivated. This can help to understand how they might have adapted to different environmental conditions, potentially revealing which genes have been expressed, if paired with meta/transcriptomics. We used culture-dependent methods in this way to understand how P. camemberti adapts to different plant cheese substrates, which revealed quite distinct morphotypes between dairy and soy substrates. These methods can be useful especially when morphological changes have occurred without genetic adaptation—another example of how understanding DNA, while powerful, can never tell the whole story.
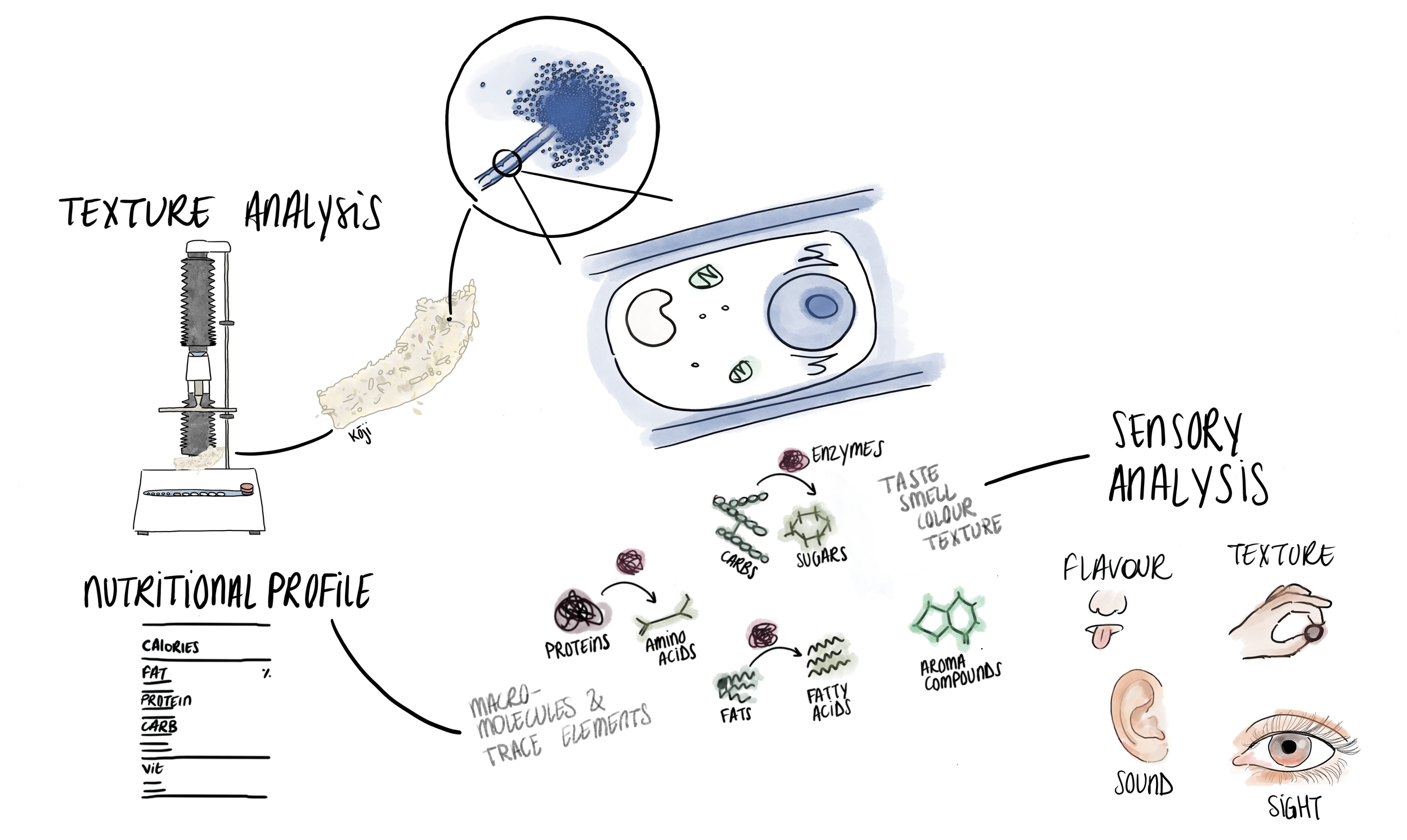
How do the microbes impact a food's taste, smell, texture, and nutritional profile?
Sensory analysis is another technique that is essential for our flavour-led approach to food innovation and research. Rather than detecting the presence of specific genes or compounds, it uses human perception as a tool for describing the taste, smell, texture, appearance, mouthfeel, and even the sound of a food product, all of which impact food acceptance as it shapes and is shaped by food culture.⁴
‘Subjective’ tests can be used to measure acceptance, preferences, and liking, for example by rating foods on a hedonic scale. Example questions might include ‘how much do you like the texture/aroma/flavour of this product—from “extremely dislike” to “extremely like”?’ or ‘would you prefer this food product if it were sweeter?’ Questions like the latter can be used to adjust the product’s sensory characteristics. ‘Objective’ tests can be used to measure sensory properties. This requires training a panel of people to establish a shared, calibrated perception of specific taste, aromatic, and/or textural properties, who can then test and describe these qualities in specific products, for example the intensity of bitterness in beer.
We combined these methods with metabolomics in the space miso project to get a fuller picture of the flavours people perceived and to connect these percepts to how the flavours had formed chemically. For example, participants identified nutty and roasted aromas in the space miso that were reviewed positively in consumer analysis. These aromas were linked with the greater presence of pyrazines in the space miso, compounds produced through the Maillard reaction which is accelerated at higher temperatures. We used a similar combination of volatile aroma compound analysis (a kind of metabolomics) and sensory analysis in our collaborative project on insect frass tea, to link its flavour chemistry with how its flavours were perceived.
Using texture analysis we can measure parameters such as hardness, cohesiveness, adhesiveness, viscosity, elasticity and chewiness to understand a food's textural attributes, another important factor in food acceptance. In our soy cheese model research, we coagulated soy with different coagulants and used a texturometer to analyse the differences in texture provided by each coagulant. We might also use this method in our R&D work with tempe.
Nutritional analysis can be used to better understand the nutritional profile of new foods. Analysis of essential nutrients like proteins, carbohydrates, fats, vitamins and minerals can help us understand how a product fits into different dietary requirements. It can also be used to optimise production to ensure that the final product is as nutritious as possible alongside other important characteristics like flavour and texture. Standard laboratory protocols exist to reveal the general macro- and micronutrient content of foods, like dissolving a sample in strong acid and quantifying the nitrogen released (the Kjeldahl method) to measure its protein content.⁵ Methods like LC-MS, GC-MS, and even microscopy can all be used for more detailed nutritional analysis to reveal specific compounds.⁶
One example of where these methods are useful in our work is in ensuring any plant-based analogues have comparable nutritional value to their animal-based counterparts. High-quality animal products from healthy agroecosystems are very nutrient-dense (partly why they are so delicious), and plant cheeses based on translated traditional techniques would ideally be similarly nutrient-dense, rather than ultra-processed products with little nutritional value. We reviewed the fatty acid profiles of plant and dairy cheeses currently available on the market and found that whilst they have similar lipid content, they have significantly different fatty acid profiles. The plant-based cheeses are rich in mono- and polyunsaturated fatty acids, such as oleic and linoleic acids, respectively, while dairy cheeses have more saturated fatty acids. This knowledge can in turn guide our R&D process in future. We also used nutritional analysis during our research on the enhancement of the microbial production of vitamin B12—a key nutrient rare in plant-based foods—in the fermentation of plant cheese. Similar methods will also be useful in all kinds of upcycling experiments, to ensure that the products are not just fun and novel but also nourishing, an important consideration when using by-products and sidestreams.
iii. Future research
Overall there is a wealth of scientific methods available to us to better understand food and facilitate flavour-forward innovation. Some of these methods we’ve used a lot; others we hope to explore more in the future.
We are always excited to explore collaborative research projects around food, fermentation, and flavour. If you have a project in mind that uses any of these scientific methods, feel free to reach out to Josh or Caroline.
Contributions & acknowledgements
Eliot wrote the article in discussion with Caroline, with contributions from Nabila. Josh provided additional written contributions and editorial feedback.
Sara Vande Velde made the illustrations of the scientific methods.
Related posts



Endnotes
[1] Patrick Schloss and Jo Handelsman (2003), ‘Biotechnological prospects from metagenomics’, Current Opinion in Biotechnology; however, some researchers too readily discard culture-dependent methods, which still have many uses, according to: Connie Ha and Susanne Devkota (2020), ‘The new microbiology: cultivating the future of microbiome-directed medicine’, American Journal of Physiology.
[2] The suffix ‘-ome’ in molecular biology refers to the total set of the thing the suffix modifies—so ‘genome’ refers to the total set of all genes in an organism, ‘metagenome’ refers to the total set of all genomes in a sample, ‘metabolome’ refers to all metabolites in a sample, etc. The suffix ‘-omics’ refers to the method/s used to analyse these total sets. See more in Jan David Brüwer and Hagen Buck-Wiese (2018), ‘Reading the Book of Life – Omics as a Universal Tool Across Disciplines’, in: Simon Jungblut, Viola Liebich and Maya Bode (eds), ‘YOUMARES 8 – Oceans Across Boundaries: Learning from each other. Proceedings of the 2017 conference for YOUng MARine RESearchers in Kiel, Germany’.
[3] Petr Kralik and Matteo Richi (2017), ‘A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything’, Frontiers in Microbiology.
[4] Claudia Ruiz-Capillas and Ana M. Herrero (2021), ‘Sensory Analysis and Consumer Research in New Product Development’, Foods.
[5] Maria Hayes (2020), ‘Measuring Protein Content in Food: An Overview of Methods’, Foods.
[6] Some of the techniques (and their protocols) that can be used to assess the nutritional profile of foods are detailed in Deb Duhita Mondal et al. (2023), ‘An overview of nutritional profiling in foods: Bioanalytical techniques and useful protocols’, Frontiers in Nutrition.